Research
Our overarching goal is to explore and understand how biomolecules (e.g., proteins and RNA) reach their functional native states starting from an astronomical number of random coil conformations.
Our goal is to primarily focused on discovering general principles that govern the folding of biomolecules. We use a combination of theoretical and computational strategies to answer several questions. In particular, we are interested in:
- RNA tertiary structure prediction
- RNA 3D structure evaluation
- Folding mechanisms of large RNA and DNA molecules
- Data-drive approaches to RNA computational biology
- AI-based RNA design
Here are some themes and techniques that we currently work on:
RNA coarse-grained modeling We have developed a novel coarse-grained model for RNA tertiary structure prediction. The model can predict 3D structures, thermodynamical stability, and flexibility of RNAs including pseudoknots and junctions in monovalent and divalent ion solutions only from single sequence.
RNA 3D structure evaluation We have developed an all-atom distance-dependent statistical potential based on residue separation for RNA 3D structure evaluation, namely rsRNASP, which is composed of short- and long-ranged potentials distinguished by residue separation. The extensive examinations against available RNA test datasets show that rsRNASP has apparently higher performance than the existing statistical potentials for the realistic test datasets with large RNAs from structure prediction models, including the newly released RNA-Puzzles dataset, and is comparable to the existing top statistical potentials for the test datasets with small RNAs or near-native decoys. In addition, rsRNASP is superior to RNA3DCNN, a recently developed scoring function through 3D convolutional neural networks.
AI for RNA 3D structures.
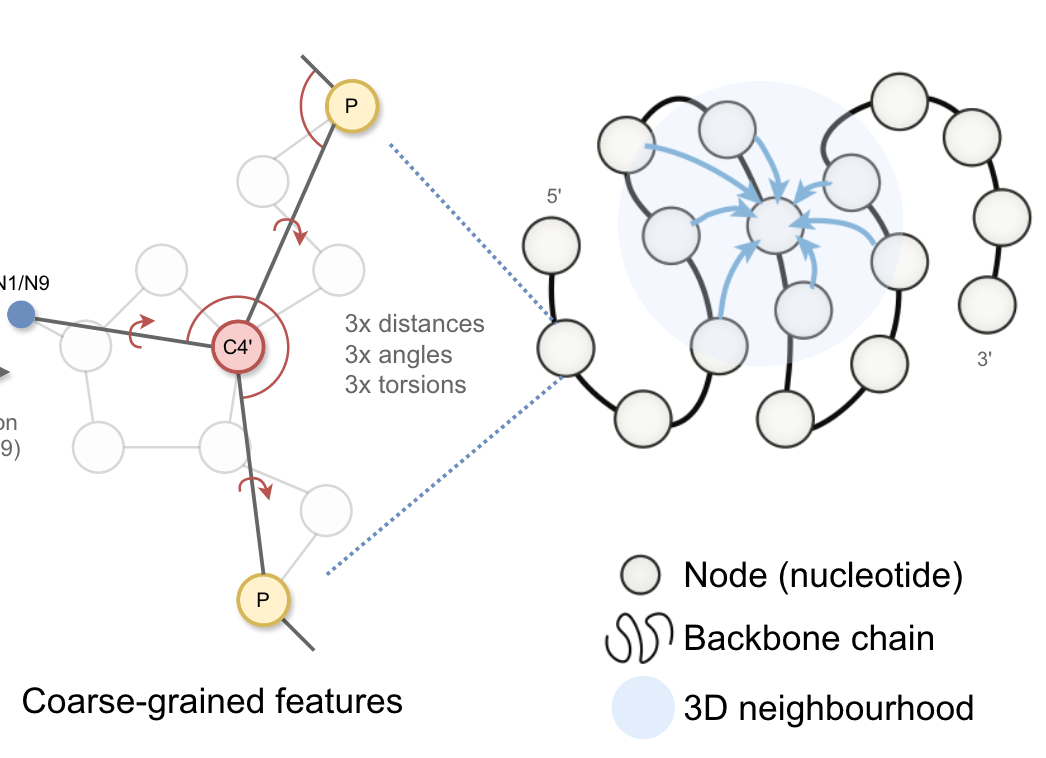 One of the projects back from our job-proposal is to develop AI models such as Graph Neural Network for RNA 3D structure characterization, based on which we can combine our physical models to predict RNA 3D structure from sequence or design RNA sequence from bacbone structures.
One of the projects back from our job-proposal is to develop AI models such as Graph Neural Network for RNA 3D structure characterization, based on which we can combine our physical models to predict RNA 3D structure from sequence or design RNA sequence from bacbone structures.
We also focus on RNA interactions with proteins, small molecules, and ions, as well as complex structure prediction.